 Home
HomeOfficial Journals By StatPerson Publication
|
Table of Content-Volume 5 Issue 2 February 2018
Genetics in periodontics: An overview
Esha Tyagi*, Tanushree Bera, Vikas Kacran, Rinki Tripathi, Pragya Jha
Department of Periodontics, Army College of Dental Sciences, Rajiv Swagruha ABHIMAAN Project, Secunderabad, Telangana 500083 Email: eshashimmer@gmail.com
Abstract Periodontal disease was earlier believes to be a single disease with variations in clinical symptoms. However, extensive researches over a long period of time have now established that periodontitis is actually a multifactorial and a bidirectional disease with genetic predisposition to it playing an intergral role. Hence, it is important to consider known risk factors for periodontal disease when focussing on familial clustering or possible genetic mechanisms, as many risk factors for periodontal disease tend to cluster in families through genetic or culture mechanism.. Identification of such genes could enable clinicians better to identify high-risk individuals for targeted prevention and treatment. The more susceptibility factors an individual has inherited, the greater the genetic predisposition and the higher the chance for early development of periodontitis. Hence, this review focuses on the detailed interplay between genes and periodontitis to provide a better understanding for the same. Key Words: periodontics.
GENETICS is the study of the inheritance, or heredity of living things. It is a wide ranging science that explores the transmission of biological properties (traits) from parent to off spring. Many human diseases are influenced by heritable alterations in the structure or function of genes. The term ‘genetics’ was coined by Bateson in 1906 What is a gene? (Johannsen 1909) Gene- a Greek word meaning “to become” A hereditary unit that occupies a specific position within a genome or chromosome that has one or more specific effects upon the phenotype of the organism. Human genome contains 25,000 to 35,000 genes. DNA - DEOXYRIBONUCLEIC ACID -blueprint of life (has the instructions for making an organism) It was established by James Watson and Francis Crick
Figure 1: Figure 2:
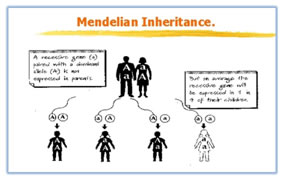
GENOME: A term used to refer to all the genes carried by an individual or cell. We have approximately three billion base pairs (6 billion bases total) of DNA in most of our cells. Terminology: Genotype: The genetic makeup of an organism or cell distinct from its expressed features. Phenotype: The observable characteristics displayed by an organism as influenced by environmental factors and is dependent on the genotype. Locus: The physical location a gene occupies within a chromosome or portion of genomic DNA. Allele: Alternate forms of a given gene differing in DNA sequence assumed to arise by mutation and affecting the function of a single product. Linkage: The tendency for certain genes to be inherited together due to their presence on the same chromosome. Linkage disequilibrium: Occurrence of some genes together more often than would be expected by random distribution. Genetic marker: refers to any gene or nucleotide sequence that can be mapped to a specific location or region on a chromosome. Penetrance: refers to the probability that a particular phenotype will result from a genotype. Partially penetrant: means that only a fraction of individuals who inherit the disease alleles will be affected. Mutation: A mutation is a permanent change in the DNA sequence of a gene. Mutation in a gene’s DNA sequence can alter the amino acid sequence of the protein encoded by the gene. Heterogeneity: means that there are different causes of disease among families. Concordance: level of similarity in disease experience. Mendelians Laws
It is now evident that there is a genetic basis for most diseases including periodontitis. Elucidation of the genetic basis of periodontitis should permit a better understanding of disease etiology, allowing improved classification, diagnosis, and treatment of periodontal diseases. Thus there has been enormous effort expended so far to identify gene polymorphisms associated with the risk for the periodontal diseases. Reports of genetic polymorphisms associated with periodontal disease are increasing, yet the limitations of these reports are not widely recognized. To appreciate these limitations it is necessary to understand the genetic basis of complex diseases. It is increasingly evident that genetic variance is a major determinant of the differential risk for many human diseases. While microbial and other environmental factors initiate and modulate periodontal disease, individuals are known to respond differently to common environmental challenges, and this differential response is influenced by the individual's genetic profile. Genes clearly play a role in the predisposition to and progression of periodontal diseases. Support for the idea that genetic factors are important determinants of periodontitis susceptibility and progression comes from studies of humans and animals which indicate that genetic factors which impair inflammatory and immune responses in general, affect periodontitis experience specifically. It is hoped that identification of the specific genes causing periodontal disease susceptibility may have diagnostic and therapeutic value. To understand the potential clinical relevance of genetic variability on periodontitis it is necessary to understand how different genes can contribute to disease. There are estimated to be 20,000-25,000 different genes in the human genome. Genes can exist in different forms or states. Geneticists refer to the different forms of a gene as allelic variants, oralleles. Genetic alterations could also change the transcript level of the protein, that is, the polymorphism may occur not in the protein coding region but in the gene promoter region, and thus influence the amount of protein produced by the gene. Allelic variants of a gene differ in their nuc-leotide sequence. Different allelic forms of a gene can produce an identical protein or different iso-forms of a protein. In the former case, the genetic polymorphism does not change the amino acid and the genetic change is said to be 'silent'. In the latter case, a nucleotide change alters the amino acid composition of a protein. The results can range along a continuum of functional consequences, from no observable change in protein function, to a minor change in function, to a dramatic change or obliteration of function. Individual genetic changes that are causal of disease are typically the result of a genetic alteration that dramatically alters a protein's function. Such genetic changes are typically termed mutations and are rare on a population level, typically being present in less than 0.1% of the population. When a specific allele occurs in at least 1% of the population, it is said to be a genetic polymorphism. In contrast to mutations, these more common genetic polymorphisms are usually considered normal variants in the population. Common genetic polymorphisms may change the function of a protein, but usually the change is relatively minor. Consequently, the specific protein products of different alleles may function differently. These differences in physiological functioning of different proteins can be enhanced by certain environmental exposures, e.g. diet, smoking, or microbial factors. If the affected protein functions in a biological process, e.g. inflammatory response to a specific microbial agent, certain polymorphisms may increase or decrease a person's risk for a disease phenotype. Whether a particular gene variant contributes to a disease phenotype depends upon the magnitude of the effect in contributing to the development of disease.
SIMPLE MENDELIAN' DISEASES
Figure 3:
Diseases that follow predictable and generally simple patterns of transmission have been called 'Mendelian' conditions. The name reflects the fact that these diseases occur in simple patterns in families and in most cases genetic alterations at a single gene locus are the major determinant of the clinical disease phenotype. These diseases follow a classic Mendelian mode of inheritance (autosomal dominant, autosomal recessive or X-linked). The disease phenotype usually manifests over a broad range of environments, and although environmental factors and other genes can modify the clinical presentation, in most cases the mutation will manifest in a remarkably similar phenotype. The population prevalence of individual Mendelian diseases is rare (typically much less than 0.1%), with the exception of some unique populations that have been isolated from other human populations. Examples of Mendelian type diseases include amelogenesis imperfecta, Crouzon syndrome, cleidocranio dysplasia, and Papillon-Lefevre syndrome (Table 1). When the gene responsible for a Mendelian disease has been identified, it is often possible to develop a diagnostic test to identify individuals who carry a disease-causing mutation in the responsible gene.
Table 1:
COMPLEX GENETIC DISEASES Genetically complex diseases differ from simple Mendelian diseases in several important ways. Genetically complex diseases do not follow a simple pattern of familial distribution or transmission. In contrast to 'simple Mendelian traits', which are caused by a single gene, these 'complex traits' are the result of the interaction of alleles at multiple different gene loci. Environmental factors are usually etiolog-ically important, and often necessary, in the development of complex diseases. On a population level, complex genetic diseases are much more common than simple Mendelian diseases, and many occur with a population prevalence of greater than 1%. Polymorphism Vs. Mutation: A major difference in the genetic basis for simple Mendelian diseases vs. complex genetic diseases is the number of genes involved and the contribution of each gene to the overall disease phenotype. In Mendelian disease, alteration of a single gene locus can result in disruption of a protein product that has a major physiological impact and therefore may be considered to be deterministic of the disease. The fact that the genetic alteration is predictably associated with a disease phenotype indicates that there is no redundancy or compensation in the particular biological system that can overcome the effect of the underlying genetic defect. In contrast, the genetic alterations that contribute to complex diseases are individually responsible for much more subtle perturbations of protein function. There is not a one-to-one correlation of the presence of a specific genetic allele and the occurrence of a complex disease, but rather specific alleles are reported to be found more frequently in diseased individuals than in nonaffected controls. Often initial support for the link is the statistical 'association' of an allele with a disease state. It is also important to understand that disease alleles reported to be associated with a complex disease are also found in unaffected individuals. Because multiple different gene alleles can contribute to a complex disease state, an individual with a complex disease need not have all of the alleles reported to be associated with that disease. Thus, the presence of a disease-associated allele in an individual is not diagnostic for a disease. Environmental factors are also critical to the etiology of most common diseases. Most chronic diseases are of adult onset, and therefore take many years to develop. Genes involved in the innate, inflammatory, and immune responses are often involved. These physiological and biological pathways have many compensatory and redundant aspects, and therefore it is extremely difficult to quantify the effect of any one genetic variant to the disease state.
Figure 4:
Single Nucleotide Polymorphisms: The most common type of genetic polymorphism is a Single Nucleotide Polymorphism (SNP). There are an estimated 10 million SNPs in the human genome. SNPs occur throughout the genome, in regions encoding genes as well as in regions without identified open reading frames. SNPs in genes may occur in protein coding regions (exons) and noncoding regions (introns and regulatory regions). Many SNPs that occur in genes have no effect on the encoded protein, but a large number of SNPs do have an effect on the gene product. Whether this change contributes to a disease phenotype depends on the specific consequence of the genetic variant on the function of the gene and its protein product. How Different Genes Can Contribute To Disease
Figure 5:
 Methods of Genetic Analyses Familial Aggregation: Familial aggregation of a trait or disease can suggest genetic etiology. However, families also share many aspects of a common environment, including diet and nutrition, exposures to pollutants, and behaviors such as smoking (active and passive). Certain infectious agents may cluster in families. Thus, familial aggregation may result from shared genes, shared environmental exposures and similar socio-economic influences. To determine the evidence for genetic factors in familial aggregation of a trait, more formal genetic studies are required TWIN STUDIES: Through the phenomenon of twins, in particular monozygous twins, who arise from one fertilized egg, nature has provided a wonderful tool for the examination of genetic influences in disease and for partitioning the relative contribution of genes and environment to a trait. Monozygous twins are genetically identical. Dizygous twins are only as genetically similar as brothers and sisters would be, on average they share ~ 50% of their genes in common (dizygous twins are from two different eggs and two different sperm). Discordance or differences in the presence of disease between monozygous twins must be due to environmental factors. Disease discordance between dizygous twins could arise from both environmental and genetic differences. Segregation Analysis: Genes are passed from parents to children in a predictable manner, and usually segregate in families as predicted by Mendel's laws. Geneticists can study the pattern of disease transmission in families using a method called segregation analysis. Segregation analysis evaluates the relative support for different transmission models to determine which model can account for the observed segregation of a trait through families. By sequentially comparing models to each other, segregation analysis identifies the model that best accounts for the observed transmission of a trait in a given population. Geneticists generally apply segregation analyses to determine whether a trait transmission appears to fit a Mendelian or another mode of genetic transmission. Segregation analysis tests alternative models to try to develop the best characterization of transmission characteristics within a set of data. As such, it is most appropriately applied to data sets of many families to determine the best fitting model. Segregation analysis does not find or aim to find a specific gene responsible for a trait.
Figure 6:
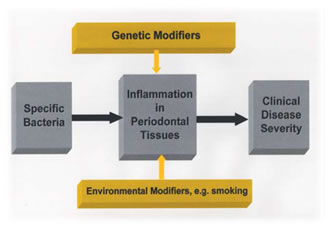
Linkage Analysis: Linkage analysis is a technique used to localize the gene for a trait to a specific chromosomal location. Genetic linkage studies are based on the fact that alleles at syntenic gene loci in close proximity on the same chromosome tend to be passed together from generation to generation (i.e. segregate), as a unit. Such genes are said to be 'linked', and violate Mendel's law of independent assortment. Geneticists can apply quantitative analyses to detect this lack of independent assortment of genetic loci, and can use it to map (localize) genes to specific chromosome locations. Over the past 15 years, genetic maps have been developed that show the position of millions of polymorphic genetic loci spanning the human genome. Scientists can follow a specific trait as it segregates through families of interest and determine whether the trait appears to segregate with a known genetic polymorphism that has been localized to a specific chromosomal location. In this manner, scientists can test whether a trait appears to segregate in a manner consistent with 'linkage' to a known genetic marker. Because the precise chromosomal location of the genetic marker is known, when linkage is detected, the gene responsible for the trait can be placed in the vicinity of the linked genetic polymorphism. Linkage can therefore prove the genetic basis of disease. Linkage is often used as a first step to determine the approximate location of a gene of interest, permitting subsequent studies to identify the mutation responsible for a disease trait. Association Studies: Genes contributing to common, complex diseases such as periodontitis have proven more difficult to isolate. When multiple, perhaps many, genes act with environmental factors to contribute to disease liability, it is difficult to formulate disease models. In the absence of specific genetic models, the etiology of complex diseases is often conceptualized as due to multiple factors, i.e. several genetic loci interacting with each other to produce an underlying susceptibility, which in turn interacts with additional environmental factors to produce an actual disease state. Theoretical research suggests several reasons for the ambiguity of the linkage results in these cases. First, if a disease gene is neither necessary nor sufficient to cause a disease, but rather is a 'modifier gene' that elevates a nonzero baseline risk, conventional parametric linkage analysis may not detect the gene. Second, if the relative contribution of a gene to a disease phenotype is small, i.e. the disease susceptibility allele raises the risk by a factor of <2, linkage analysis using affected sibling pairs will not be powerful enough to detect the gene, given realistic sample sizes. Thus, linkage analyses may not be a useful strategy to detect modifier genes or genes that exert small effects - precisely those genes which might be operating in chronic periodontitis and many other complex disorders. International Hapmap Project: The international HapMap project is being conducted to identify and catalog the common genetic variants that occur in human beings. The project will describe each of the common SNPs in the human genome. The goal is to determine the genetic location of each SNP and characterize how these genetic variants are distributed in several different population groups. The project is at present studying DNA samples representative of African, Asian, and European ancestry. By identifying most of the 10 million SNPs that occur in humans, the HapMap project will identify the DNA variants responsible for most of the genetic diversity in humans. The project is not intended to identify specific disease-associated polymorphisms. Instead, it is hoped that by providing such a catalog of common genetic variants, clinicians and scientists can work together to identify SNPs with important disease associations to understand disease etiologies and develop new diagnostic and treatment strategies. Evidence for The Role of Genetic Variants in Periodontitis: The risk for many diseases, including periodontal diseases, is not borne equally by all individuals in a population. A variety of microbial, environmental, behavioral, and systemic disease factors are reported to influence the risk for periodontit. An individual's genetic makeup is a crucial factor influencing their systemic or host response-related risk. There are reports in the literature on familial aggregation of periodontal diseases, but it is difficult to compare them. Although periodontal disease terminology has changed many times over the years, most familial reports of early onset forms of periodontitis are now referred to as aggressive periodontitis. Reports of the familial nature of chronic forms of periodontitis are less frequent but the aggregation within families is consistent with a genetic predisposition. We should remember, however, that familial aggregation of periodontal disease may also reflect exposure to common environmental factors. Shared environmental factors include education, socioeconomic grouping, oral hygiene, shared transmission of bacteria, diseases such as diabetes and environmental features such as passive smoking, sanitation, etc. Some aspects of behavior are determined in part by genetics, which may influence potential modifying factors such as education, lifestyle and, of dental importance, oral hygiene. Complex interactions between genes and environment are difficult to quantify, but are likely to be important when considering the familial risk for the periodontal diseases. In chronic periodontitis, clinical disease characteristics do not present until the third decade of life, whereas in the aggressive forms of periodontal disease, the presentation can occur much earlier, in the early teens or younger. This variability in presentation makes diagnosis difficult, not only in declaring disease but also in detecting patients who are free of the disease and in differentiating between chronic and aggressive forms of periodontitis. The problems associated with the clinical diagnosis of periodontal disease are not uncommon in medical genetics as similar problems arise when studying other delayed onset genetics. The problems of genetic model testing in aggressive periodontitis have been highlighted by Boughman et al. who noted that aggressive periodontitis has a variable age of onset and is often not recognized until after puberty. Diagnosis of aggressive periodontitis in older individuals is problematic due to the difficulties of distinguishing between aggressive periodontitis and chronic adult onset forms of periodontitis on the basis of the clinical signs. Similarly, diagnosis in older edentulous individuals is also problematic. The effects of the environment, for example plaque accumulation and smoking, also have major long-term influences on disease experience (Fig. 1) and these confound the diagnosis of aggressive periodontitis. Linkage Studies In Aggressive Periodontitis: Three linkage studies have been performed to date on families with localized aggressive periodontitis.Boughman et al.14 identified an autosomal dominant form of localized aggressive periodontitis in an extended family from Southern Maryland. In this family, type III dentinogenesis imperfecta and a localized form of aggressive periodontitis were segregating as dominant traits. Since the gene for dentinogenesis imperfecta-III had been previously localized to chromosome 4, they performed a linkage analysis on this chromosome and demonstrated a relatively close linkage with the suspected locus for aggressive periodontitis14. This was an important study because it supported autosomal dominant inheritance of a single major gene locus, clearly indicating a major genetic component to the aggressive periodontitis disease etiology. Hart et al.69 evaluated support for linkage to this region of chromosome 4 in a different population of families (14 African American and 4 Caucasian). Their findings supported genetic locus heterogeneity of aggressive periodontitis, as they excluded a chromosome 4 major gene locus for aggressive periodontitis in the families they studied. Thus, this Brandywine population appears to have a different form of periodontal disease with a different gene being responsible compared with the African-American and Caucasian families studied by Hart and coworkers. These findings support genetic heterogeneity, with at least one gene locus responsible for aggressive periodontitis located on chromosome 4. Recently, Li and coworkers107 reported evidence of a gene responsible for localized aggressive periodontitis located on chromosome 1q25. To date, a gene of major effect for aggressive periodontitis has not been identified. Syndromic Forms of Periodontitis: Significant, irrefutable clinical and laboratory data indicate that genetic variants do predispose to disease states in humans. Severe periodontitis presents as part of the clinical manifestations of a number of monogenic syndromes and the gene mutation and biochemical defect is known for many of these conditions. A commonality of these conditions is that they are inherited as simple Mendelian traits due to genetic alterations of a single gene locus. Examples of these monogenic conditions are given in Table 1. The significance of these conditions is that they clearly demonstrate that a genetic mutation at a single locus can impart susceptibility to periodontitis. Additionally, these conditions illustrate that this genetic susceptibility may segregate by different transmission patterns.
Because altered proteins function in different structural and immune pathways, genetic modulation of a variety of different genes can affect a variety of different physiological and cellular pathways. These conditions illustrate that the genetic contribution to periodontitis susceptibility is multifaceted, and may potentially involve many different gene loci. However, in contrast to non-Syndromic forms of periodontitis, these conditions have periodontal disease manifestations as part of a collection of Syndromic manifestations. In most cases of aggressive periodontitis, individuals present with clinical manifestations of periodontitis, but do not appear to have any other clinical disease manifestations. This is not inconsistent with a genetic disease etiology. Expression of genes can vary in different tissues, and mutations of a ubiquitously expressed gene can result in a tissue-specific condition. Recently, mutation of the SOS1 gene has been identified in individuals with hereditary gingival fibromatosis. SOS1 is important in determining whether cells grow, divide or differentiate and is ubiquitously expressed. However, the only clinical manifestation of this gene defect appears to be enlargement of the periodontium, an instance of the tissue-specific nature of many diseases. A similar tissue-specific manifestation of a gene defect may occur in nonsyndromic aggressive periodontitis.
Figure 7:
Gene Polymorphisms of Host Response Elements and Periodontitis: The genetic basis of many aspects of the periodontal host response has been discussed in reference to genetic disorders predisposing to periodontal disease. The aim of this section is to generally summarize the potential influence of innate, inflammatory, and immunological genetic variations and to consider where the most promising candidates lie from the viewpoint of a genetic diagnostic approach to periodontitis. There is a genetic basis to many aspects of the periodontal host response. Mutations of genes have been identified in Mendelian inherited syndromes that have significant periodontitis findings, clearly indicating that single gene defects can predispose to periodontitis. Data from human and animal studies indicate that genetic variance can influence the innate, inflammatory, and immunological response to microbial infection. The availability of the annotated human genome and technological advances in genotyping have made it possible to genotype allelic variants and to test for association in case-control studies. This has facilitated studies to evaluate the support for/against the association of an array of genetic polymorphisms with periodontitis periodontitis. It is important to realize that most of these studies are underpowered to draw definitive conclusions. Several features of the host's innate immune system which may contribute to genetic susceptibility to aggressive periodontitis have been outlined already and include epithelial, connective tissue, and fibro-blast defects. Functional defects or a deficient number of polymorphonuclear leukocytes, as discussed previously, have profound effects on the host's susceptibility to periodontitis. Another aspect of the host inflammatory response, pro-inflammatory cytokines, has attracted much attention as potentially crucial variants influencing the host response in periodontitis. This review will not attempt to summarize the plethora of SNP studies in periodontal disease but will summarize tabular the areas of current research and focus on the interleukin-1 polymorphism by way of example and illustration of the SNP periodontal literature. Interleukin-1 Polymorphism In Aggressive Periodontitis: Hodge et al. examined interleukin-1a and interleukin-1 b genetic polymorphisms in unrelated European white Caucasian patients with generalized early onset periodontitis and found no significant differences between patients and controls for any of the 'composite genotypes' described by Kornman et al. No significant differences were found between patients and controls whether smoking was included as a covariate or not. It was concluded that there was a lack of association between the interleukin-1 polymorphisms and aggressive periodontitis, which questions the utility of these candidate genes as markers of susceptibility. This was a relatively homogeneous Scottish population and the results, although negative, merely reflect the lack of utility of this 'composite genotype' test in this population.
Table 3: Cytokine polymorphism studies in periodontal L disease
FC, FMLP Receptor Polymorphisms: The Fc portion of an antibody is responsible for binding to receptors present on immune cells independent of the antigen-binding, Fab portion. Thesecellular Fc receptors, especially IgG Fc receptors designated FcgRs have been the subject of numerousinvestigations in periodontal research. They arefound on cells capable of binding the Fc portion ofIgG antibody such as macrophages, monocytes, and PMNs and are important in the phagocytosis, antibody-dependent cell-mediated cytotoxicity, endocytosis, enhancement of antigen presentation, and there lease of inflammatory mediators. FcgRs are currently classified into FcgRI (CD64), FcgRII (CD32), and FcgRIII (CD16) each containing is oforms within each class that affect cellular distribution and binding affinity to IgG subclasses. Polymorphisms in the genes encoding the low affinity receptors FcgRIIa, FcgRIIb, FcgRIIIa, and FcgRIIIb may resultin variations in antibody binding and phagocytosisand hence susceptibility to periodontitis. The existence of several FcgR polymorphisms of periodontal relevance has been documented andappears to be involved in microbial clearance. TheFcgRIIa-131 H/R polymorphism (histidine, H or arginine, R) at position 131 in the membrane proximal Ig-like domain results from a single G to A nucleotidesubstitution. IgG2-opsinized particles are phagocytizedmuch more efficiently in PMN’s from FcgRIIa-H/H genotypes than from FcgRIIa-R/R genotypes. The FcgRIIIa-158F/V polymorphism in the high affinityreceptor involves a phenylalanine, F to valine, Vamino acid substitution at position 158 in the Ig-likedomain as a result of a G to T substitution at nucleotide 559 in the DNA sequence. The V/V variant iscapable of efficient binding of IgG1, IgG3, and IgG4relative to the F/F variant in both monocytes and natural killer cell. This substitution was also associated with recurrence of adult period on titis compared to individuals without recurrence in Japanese patients. Implants: Implants are now an integral part of periodontal and restorative dentistry. However, due to the some what recent introduction of predictable therapies inimplant dentistry, very few studies have been reported addressing the influence of genetics onimplant survival. The effects of the IL-1 genotype, smoking status, and patient’s age on failed or failingimplants was recently compared to successfully integratedimplants. Although smoking increasedthe risk of implant failures by approximately 2.5 times, statistical testing failed to indicate a relationshipbetween implant failure, age, or IL-1 genotypestatus. This was further validated by another studythat failed to demonstrate a relationship betweenimplant failure and the IL-1A (_899), IL-1B (þ3953) composite genotype in Caucasian patients. However, it was suggested that the absence of thesealleles may not indicate a reduced risk of periodontitisor peri-implantitis.Calcitonin is a hormone produced in the thyroidthat causes a reduction of calcium ions in the blood.The relationship between the calcitonin receptorgenotype and mandibular buccal marginal bone lossat stage II surgery in 237 implants was recentlyreported in Japanese patients. Although therewere no significant differences in the distributionof age, smoking status, postmenopausal women,and bone quality, patients with the calcitonin receptorpolymorphism were 20 times more likely to sufferbuccal marginal bone loss than patients who werecalcitonin receptor genotype-negative. Smoking and Susceptibility Genes: An important environmental risk factor is smoking. The interplay between smoking and genes in the risk for periodontitis has been rightly discussed in most studies on genetic risk. Interestingly, the attributable risk for periodontitis from smoking may be influenced by the polymorphism of the N-acetyltransf-erase (NAT2) gene, which may change the subject's metabolism to produce more damaging smoking-derived xenobiotics. Meisel et al hypothesized that an NAT2 genotype could be a risk factor for periodontal disease. Severe chronic periodontitis patients were predominantly slow acetylators (OR = 2.38-5.02). Thus, the slow acetylator phenotype may be associated with a higher risk for periodontitis. This is an example of a strong gene-environment interaction. Biologic Plausibility: Researchers should always consider the biological plausibility of the effect of the gene polymorphism in the periodontal disease process. There are, however, a great many unknown aspects which provide considerable leeway for investigators in this area. The pathogenesis of periodontal disease is to some extent known. It is a chronic inflammatory condition which progresses to the destruction of bone and supporting structures and ultimately the loss of teeth, but the reason that some patients are more susceptible than others is not understood. Any of the innate, inflammatory, or immune processes may be involved in the etiology of periodontal disease and, in addition, it is feasible that structural aspects and aspects which may influence the healing process may be similarly involved. This leaves an extremely wide range of factors, which could be considered fortuitous from a research justification viewpoint but frustrating in the search for the true etiology of the periodontal diseases. The Lack Of Logic In Association Studies: A hypothetical example of the logic in play when studying gene polymorphism associations with diseases might be the following. In periodontitis (condition A), bone is lost (abnormality B), and since bone is removed by osteoclasts, which are influenced by a specific gene (gene C), we then study the association of a known polymorphism (polymorphism D) of gene C with periodontal disease. Obvious problems in this logic are apparent even superficially. Whether the osteoclast has a pivotal role in the disease process is unknown and to assume it does would be purely speculation. Nevertheless, the link between A and B typically results in the search for a gene C and a polymorphism D, which happens to be readily investigatable. Linking gene C with periodontal disease is a task for molecular geneticists, who would employ linkage analysis techniques and positional cloning together with robust statistical approaches including a precise genetic model and knowledge of the population frequency, penetrance of the disease, mutational searches, and laboratory pathogenesis investigations. Susceptibility Profiles: An interesting approach which is emerging in other fields of medicine is the development of the susceptibility profile concept in diseases such as Alzheimer's. The risk of Alzheimer's has been considered to be substantially influenced by 10 genetic polymorphisms of inflammation-related molecules including proinflammatory cytokines (interleukin-1a and interleukin-1b and interleukin-6, tumor necrosis factor-a) and the protease inhibitors (a2-macroglobulin, a1-antitrypsin). McGeer et al. (116) consider that several of these relatively common high risk polymorphisms may be inherited by an individual, giving them a susceptibility profile which reflects the combined influence of the high risk polymorphisms. A similar high risk profile may yet emerge for periodontal disease but clearly much testing across different populations will be needed. If such information was available, therapeutic intervention strategies could be envisaged, aimed at preventing the development of periodontal disease.
Genetic Screening For Periodontitis Risk: The current practical clinical utility of genetic knowledge in periodontics is limited. However, performing clinical periodontal assessments of siblings of aggressive periodontitis probands is one of the most useful actions we can perform to ensure the early diagnosis of this disease. By careful clinical diagnostic procedures we may detect susceptible patients early and instigate therapy to prevent significant disease occurring. In the pursuit of better genetic diagnostic tests for chronic and aggressive periodontitis we must plan our research using plausible biological arguments and carefully avoid bias and misinterpretation of genetic associations with disease.
CONCLUSIONS We have reviewed the fundamental differences between simple Mendelian and complex genetic diseases, and highlighted the significance of these differences for genetic testing. Simple or Mendelian genetic traits generally occur when a single gene defect disrupts the normal function of a protein sufficiently to cause a disease or syndrome. Complex genetic diseases occur when allelic variants of multiple different genes act synergistically with environmental factors to increase or decrease the likelihood of developing a disease. While there have been dramatic successes in identification of mutations responsible for rare syn-dromic forms of periodontitis, few genetic polymorphisms reported for more complex genetic forms of periodontitis have been demonstrated to be clinically valid or to have clinical utility. A review of genetic associations reported for more common forms of periodontitis reveals that we are still some way from determining the genetic basis of either aggressive or chronic periodontitis. We have, however, gained some insight into the hereditary patterns for aggressive periodontitis, which in the US population is typically inherited as an autosomal dominant trait with reduced penetrance. The clinical message here is that the risk for offspring and siblings of patients affected with aggressive periodontitis approaches 50%. As the number of reports describing genetic polymorphisms associated with periodontal disease increase exponentially, the limitations of such studies need to be appreciated. There is often a large disparity in the significance placed on genes and genetic polymorphism associations between geneticists and clinicians. A strong plea in the development of better genetic diagnostic tests for chronic and aggressive periodontitis is that we plan our research using plausible biological arguments and carefully avoid bias and misinterpretation of genetic associations with disease. Many studies fail to quantify meaningfully the magnitude of contribution of a particular disease-associated allele to disease risk. This failure to consider the sensitivity and specificity of these tests precludes determination of the clinical utility of the association. Additionally, environmental confound-ers are often not adequately addressed. Underpowered studies have insufficient numbers of cases and controls to make robust pronouncements on associations. Rarely is a precise account of race and ethnicity for cases and controls included. Biases can occur in multiracial areas of different socioeconomic classes, making the inference from such studies questionable. Ultimately, development of a susceptibility profile concept for complex diseases may emerge as a clinically valid approach to using genetic information for periodontal diseases. For example, a number of relatively common SNPs could be demonstrated to contribute to a high susceptibility profile. It is likely that environmental factors (smoking, microbes, dietary components) would be included in the profile assessment. To prove such a high risk profile in periodontal disease requires much testing across different populations. Such genetic information would be invaluable in therapeutic intervention strategies aimed at preventing the development of periodontal disease.
REFERENCES
|

 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.