 Home
Home
|
Table of Content - Volume 20 Issue 2 - November 2021
Prader-willi syndrome
Korrapati Aparna1*, Mrunalini Kulkarni2
1Intern, Associate Professor, Department of Paediatrics, Bharati Vidyapeeth(Deemed To Be) University, Sangli, Maharashtra , INDIA. Email: appu.korrapati@gmail.com
Abstract Background: Prader willi syndrome is a multi-systemic genomic imprinting disorder caused by lack of expression of genes on paternally inherited chromosome 151 Here, we present a case of PWS in a 5-year-old male child who had presented with excessive sleepiness and rapid breathing. Keywords: Prader willi syndrome, genomic imprinting, chromosome 15
INTRODUCTION PWS is a multi-systemic complex genetic disorder. It occurs because of absence of expression of paternally inherited genes in 15q11-q13 region. It may be due to paternal deletion, maternal uniparental disomy, imprinting defect.1,2
CASE PRESENTATION A 5-year-old male child born as1st issue of nonconsanguineous marriage. presented with excessive sleepiness and loss of appetite for 7 days and rapid breathing in the last 3 days. Child developed excessive sleepiness associated with loss of appetite in the last 7 days and rapid breathing for 3 days. Past History Child was diagnosed with PWS at the age of 6 months by DNA methylation analysis technique. Child was hospitalized multiple times for lower respiratory tract infection. Birth history LSCS done due to non-progression of labour and his birth weight is 2.9kg. No history of NICU admission. Developmental History: Mild intellectual defect
Table 1: Anthropometry
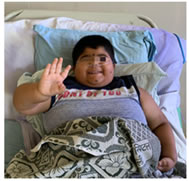
On Examination Patient was afebrile with heart rate of 170/min, RR of 50/min, and BP of 90/60 mmhg. Mild pallor and edema present. Spo2 was 78% at room air and 97% with oxygen by mask. Short stature, microcephaly, short limbs and neck RS: bilateral vesicular breath sounds heard. Breath sounds decreased bilaterally at inframammary and mammary areas. Figure 1: morbid obesity
Lab Investigations: CBC done with Hb-9.8, wbc- 16,300, platelet- 2,82,000. LFT done with SGOT (60), SGPT (552) raised, Sr protein, Sr bilirubin, RFT, Electrolytes are normal. TFT was normal. Lipid profile: Triglyceride -88(raised), Cholesterol -95, HDL - 36, VLDL -17.6, LDL -41.4, LDL/HDL= 1.15, Chol/HDL = 2.64. Chest X-Ray done showing highly placed diaphragm. USG Abdomen and Chest showed no pathology 2D-ECHO: moderate to severe tricuspid regurgitation with RVSP-68mmhg, RA/RV dilated, mild pulmonary regurgitation LVEF-60%. Pulmonary artery: moderate PAH. Diagnosis: known case of Prader willi syndrome with secondary pulmonary hypertension, central sleep apnea, morbid obesity. Treatment: Oxygen by Prong. Tab ALDACTONE 25mg Tab AMBRISENTAN 5mg. Syp TONOFERON 5ml. CPAP machine advised during sleep. Advice: Regular Physiotherapy, Dietary control to decrease obesity.
DISCUSSION PWS is a highly variable genetic disorder affecting multiple body systems. PWS was first discovered by prader et al. in 19566 and now recognised as genomic imprinting disorder. Estimated prevalence of PWS is 1/10,000 to 1/30,0001. The errors in genomic imprinting, which occur during both male and female gametogenesis, depending on the parent of origin, the loss of expression of paternal genes on chr15q11-q13 region leads to PWS and maternal genes in this region leads to Angelman syndrome, which is an entirely different clinical disorder.7,8,9 DNA methylation analysis is the technique to diagnose and differentiate PWS from Angelman syndrome. PWS is most common syndromal cause of life threatening obesity and first recognized disorder related to genomic imprinting in humans7 .Mothers often have decreased fetal activity and mostly baby found in breech positional the time of delivery2. Early diagnosis is important for effective long term management. Clinical manifestations change with age. Most common manifestations include Hypotonia and poor suck resulting in failure to thrive during infancy. As the individual ages, other features such as food seeking with excessive weight gain, unique behavioural characteristics and cognitive impairment become evident1,2,3. The phenotype is likely due to hypothalamic disfunction which is responsible for hyperphagia, hypersomnia, temperature instability, high pain threshold and multiple endocrine abnormalities include Growth hormone deficiency causing short stature for family, TSH deficiencies and hypogonadism. Obesity and its complications are the major cause of morbidity and mortality in PWS31 Management is symptomatic and supportive. Parental education about appropriate parenting techniques (structure and consistency) emphasising control of food intake and behaviour management which correlates with prognosis.4,5 Hormone replacement therapies like GH treatment in early age improves growth, body composition and muscle strength4.
REFERENCES
Policy for Articles with Open Access
|
|
 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.