 Home
HomeOfficial Journals By StatPerson Publication
|
Table of Content - Volume 9 Issue 3 - March 2019
Rare case report of Laurence-Moon-Bardet-Biedel syndrome
Divyashree P1, Manu M A2*, Rekha T D3, Dakshayani B4, Sarala Sabapathy5
1,2,3Post Graduate, 4Assistant professor, 5Professor and HOD. Department of Pediatrics, Bangalore Medical College and Research institute, Bangalore, Karnataka, INDIA. Email: manu.fulloflife@gmail.com
Abstract Laurence-Moon-Bardet-Biedl (LMBB) syndrome is a rare autosomal recessive disorder characterized by structural and functional abnormalities of different organ and tissues. In 1920 Bardet described a female patient of 4 years old with the rodcone dystrophy, marked central obesity, polydactyly and mental retardation. Biedl studied two cases in 1992 and highlighted the complete scenario of clinical signs of LMBB syndrome which could be conceivable including skull abnormalities, anal atresia and gastrointestinal conflicts. Key Word: Anal atresia, Mental retardation, Polydactyly, Rodcone dystrophy
INTRODUCTION LMBB syndrome has characteristic pentad of retinitis pigmentosa, polydactyly, obesity, mental retardation, hypogonadism1. Some consider renal involvement as sixth component of this disorder. Clinical diagnosis is based on the presence of 4 of the 5 cardinal features. Other clinical features include speech disorder, brachydactyly, developmental delay, polyuria and polydipsia, short stature, ataxia, poor coordination /clumsiness, diabetes mellitus, left ventricular hypertrophy, hepatic fibrosis and renal hypoplasia /dysplasia2,3. Laurence and Moon described retinitis pigmentosa with short stature, hypogonadism among 4 patients in a family in 18664. Bardet in 1920 observed association of polydactyly and pigmentary retinopathy among some patients with Obesity5. Biedl in 1922 described mental retardation in this syndrome6. In 1925 Solis-Cohen and Weiss connected cases described by Bardet and Biedl to the four patients described by Laurence and Moon7.
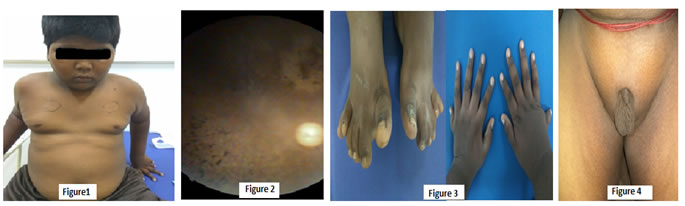
CASE REPORT A 14-year-old boy had decreased vision during night and small genitalia for past 4 years presented with increased weight gain in last 3 months with BMI of 32, born to 2nd degree consanguineous married couple with history of sibling death of unknown cause. On examination this boy had obesity, mental retardation (IQ=56), divergent squint with retinitis pigmentosa, acanthosis nigricans, polydactyly of all 4 limbs and micropenis with sexual maturity rate of stage1. Investigations revealed microcytic hypochromic anemia, hyperlipidemia, Ultrasonography of abdomen showed hepatosplenomegaly, grade 2 nephropathy with multiple renal cyst with hypoplastic prostate glands. On fundus examination he had bony specules, pale disc, attenuated arterioles suggestive of retinitis pigmentosa.
Figure 1: Central obesity, Figure 2: Fundus showing retinitis pigmentosa, Figure3: Polydactyl of all 4 limbs, Figure 4: Micropenis
DISCUSSION The patient described above had 4 typical features of LMBB syndrome with the associated clinical presentation of anemia. The syndrome is very rare, usually characterized by retinal degeneration, obesity, extra digits on the hands and feet and intellectual impairment. Modern genetic techniques have enabled the scientists to explain the syndromes by specific transformations. The detailed biochemical mechanism that leads to BBS is still unclear. Twelve genes (BBS1 to BBS12) that are responsible for the disease have been cloned. The BBS proteins are components of the centrosome and affect the ciliary transport; hence, the disease falls under the spectrum of “ciliopathies” 8. Management is supportive and includes training and rehabilitation for blind and mentally retarded patients, hearing aids for deafness, and diet and exercise for obesity. A low-calorie and low-protein diet help in obesity control and may slow the progression of renal failure in patients with LMBB syndrome9. Surgical correction can be undertaken for polydactyly. Organizations can provide emotional and social support and help in all-round development. Early and regular screening for hypertension, diabetes, and renal involvement is required10.
CONCLUSION The present patient showed all the principal features of infrequent autosomal recessive disease called Laurence Moon Bardet Biedl Syndrome. Diagnosis of this syndrome is usually missed due to its scarcity. Pediatricians and other specialties should have enough information on LMBB syndrome for accurate and spontaneous diagnosis due to its conflicting prognosis. The case is being reported for its rarity.
REFERENCES
|
|
 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.