 Home
Home
|
Table of Content - Volume 21 Issue 2 - February 2022
Intussusception in Peutz-Jeghers syndrome:A Case Report
Leander Lazarus Gomes1, Santosh Dalavi2*, Alka Gosavi3
{1Junior Resident-3, 2Associate Professor, Department of Surgery} {3Associate Professor And HOD, Department of Pathology} Government Medical College, Miraj, Maharashtra, INDIA. Email: gleanderlazarus@gmail.com
Abstract Background: Peutz-Jeghers syndrome is a rare inherited autosomal dominant disease which is characterized by the presence of mucocutaneous pigmentation over the perioral region and buccal mucosa along with multiple polyps in the gastrointestinal tract. Here we report the case of a 16-year female, who came with severe abdominal pain, vomiting, abdominal distension and history of melena. The patient also had brownish-black mucocutaneous spots over the perioral region and buccal mucosa. Ultrasonography of the abdomen and pelvis revealed the presence of a jejuno-jejunal intussusception for which laparotomy was done, and multiple polyps were visualized, the lead point of the intussusception being a polyp in the jejunum itself. The polyp was excised and sent for histopathology. Upper GI scopy and Colonoscopy also confirmed multiple polyps in the gastrointestinal tract. The patient was diagnosed with Peutz-Jeghers syndrome based on the multiple polyps in the entire gastrointestinal tract and characteristic mucocutaneous involvement. Keywords: Peutz-Jeghers syndrome, Intussusception, Mucocutaneous pigmentation, polyps.
INTRODUCTION The Peutz-Jeghers syndrome (PJS), is a rare familial disorder, in which there is an incidence of 1 in 30,000 to 1,20,000 live births.1 This syndrome is characterized by being an autosomal dominant condition with incomplete penetrance.2 This rare syndrome disorder is also characterized by the typical pigmented perioral macules, and pigmented spots in the buccal mucosa, which are present in about 90% of the patients,3,4 along with multiple hamartomatous polyps, predominantly seen in the gastrointestinal (GI) tract, rarely more than 20 in number.5 The most common location of Peutz-Jeghers polyp (PJP) is in the upper gastrointestinal tract, more specifically the upper jejunum. At times polyps may occasionally be absent also. These polyps if present vary in size from a few millimeters to upto 7cm. most of the patients depict a clinical course which characteristically shows, recurrent episodes of polyp induced bowel obstruction along with bleeding. The disease affects both males and females with equal frequency. The nonsense, frameshift and missense mutations which cause the inactivation of the LKB1/STK11 gene on chromosome 19p13.3, which codes for serine-threonine kinase, have been implicated as part of the underlying abnormality seen in Peutz-Jeghers syndrome.6 Peutz-Jeghers syndrome (PJS) presents with the characteristic flat, pigmented, and freckle-like cutaneous lesions mainly on the lower lip, perioral area, buccal mucosa, periorbital area and the eyelids. This syndrome shows a typical pathological feature which is a smooth muscle core arising from the muscularis mucosae and ramifying into the substance of the polyp like the branches of a tree. The World Health Organization (WHO) clinic-pathological criteria for the diagnosis of this rare disorder are as follows7- Three or more polyps, which show histological features consistent with PJS. A family history of PJS with any number of PJPs. A family history of PJS with characteristic mucocutaneous pigmentation. Characteristic mucocutaneous pigmentation with any number of PJPs. The individuals of PJS carry a very high risk of developing not only gastrointestinal adenocarcinoma, but also extra-gastrointestinal malignancies in the breast, pancreas, testes and ovary.8,9 As compared to the normal population, individuals with PJS have a relative risk of 15 for developing any such type of malignancy.9 In nearly 10% cases of PJS, pseudo-invasion mimicking adenocarcinoma has been described. It has been postulated that the mechanical pressure due to intussusception of small bowel polyps in PJS may be responsible for misplacing the luminal epithelial cells through the normal anatomic defects in the intestinal wall, particularly of the ones caused by traversing of the neurovascular bundles.10 Among the many published case reports with solitary or multiple PJPs, most of the patients presented with bleeding and intestinal intussusception.11,12 In all the reports of patients with single or multiple enteric intussusceptions, abdominal signs and symptoms were present to some extent. Thus this constitutes the case when presented with these clinical signs as a surgical emergency. Here we present a case of a young female, with multiple polyps present throughout the gastrointestinal tract which presented with a small intestinal intussusception (jejuno-jejunal) along with the signs of abdominal pain, distension, vomiting and history of melena.
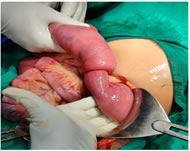
CASE PRESENTATION A 16-year old female came to our hospital with the chief complaints of with severe abdominal pain which was generalized and colicky in nature along with vomiting. Abdominal distension was also present. On examination the female had a toxic look, she was restless and also a rapid pulse was present. Severe abdominal tenderness was present in the upper abdomen, and a mass was also palpable which was mobile and firm. History of melena was also given by the mother of the patient, which was on and off. Also similar episodes of intermittent colicky abdominal pain in the past, were given by the patients mother. The patient also had history of primary amenorrhea, which was however not significant to the clinical findings. On ultrasonography of the abdomen and pelvis, a jejuno-jejunal intussusception was detected. The patient was then resuscitated, and after clinical stabilization, an emergency laparotomy was planned. During the laparotomy, it was found that about 10cm of jejunum was invaginating into the jejunum itself, which lead to the jejuno-jejunal intussusception (Fig. 1) Figure 1: Intraoperative demonstration of jejuno-jejunal intussusception
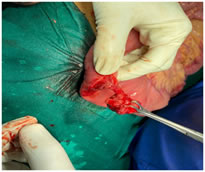
The intussusception was the reduced by milking it very carefully, and it was found out that a polyp which was present in that segment of the jejunum, was in fact the lead point of the intussusception (Fig. 2), for which an enterotomy was performed and the polyp was then removed out, and sent for histopathological examination. Figure 2: Polyp responsible for the intussusception removed through enterotomy
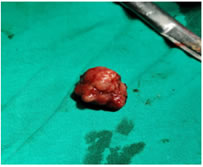
There were around 10-15 were multiple polyps, which were pedunculated on examination of the entire small bowel. The excised polyp was about 3cm in diameter, which was the lead point of the intussusception. The polyp was reddish gray in color with a smooth surface, and the cut section was congested without any focal hemorrhage and necrosis (Fig. 3).
Figure 3: Excised specimen of Intestinal polyp which was the lead point of the intussusception
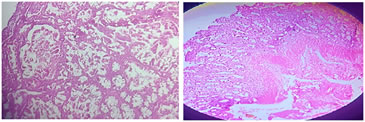
Upon histopathological examination of the specimen in was found out that changes of autolysis was present. Also polypoidal mass lined by intestinal mucosa with hyperplastic glands showing gobletization arranged in a branching pattern was seen. The core of the polyp also showed arborization and splaying of smooth muscle exhibiting Christmas tree like appearance (Fig. 4 and 5). There were areas of pseudo-invasion with entrapment of glands within the smooth muscles. The base of the polyp also showed necrotic changes. No evidence of malignancy was seen in multiple sections which were studied. These findings were suggestive of a Peutz-Jeghers polyp, based on the clinical findings as well.
Figure 4 Figure 5 Figure 4: Histopathology showing arborizing branching smooth muscle bundles between glands characteristic of PJS (H and E); Figure 5: Histopathology, showing a typical Peutz-Jeghers polyp (H and E), showing glandular disorganization, hyperplastic glands showing gobletization arranged in a Christmas tree pattern.
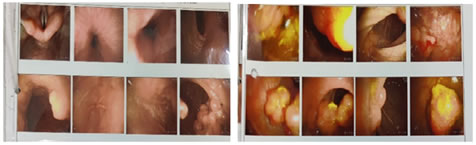
The patient upon careful re-examination had multiple pigmented mucocutaneous spots over the perioral region and buccal mucosa (Fig. 6). There was no significant family history of the patient, revealing any history of PJS/any other polyposis syndrome in the family members. Figure 6: Pigmented mucocutaneous spots seen over the perioral region and buccal mucosa On the 10th post-operative day, the female was advised an upper gastrointestinal endoscopy and colonoscopy to check for multiple polyps. The upper GI endoscopy revealed the presence of multiple polyps present at the body of stomach and D-2 and D-3 sections of the duodenum. The colonoscopy showed the presence of multiple polyps at the terminal ileum and entire colon. Figure 7 Figure 8 Figure 7: Upper GI scopy showing polyps in the stomach, duodenum; Figure 8: Colonoscopy showing multiple polyps in the terminal ileum and entire colon The patient was discharged on the 14th post-operative day with advice for regular follow-ups. On follow-up, the patient was found to be free of any complaints or any complications since the last 9 months.
DISCUSSION In 1921, Peutz published the first case with GI familial polyposis with pigmentations, which later in 1949, it was documented that these were associated with an increased risk of malignancy.3,13 PJS is the second most common hamartomatous syndrome, which occurs as an autosomal dominant condition with variable penetrance. The most distinctive clinical feature are the melanin pigmentations (black-brown spots) which can be seen in the lips and buccal mucosa. The pigmentations can also be seen in the other parts of the body, such as the fingers, toes, hands, feet, mucosa of the nose, conjunctiva and the rectum. Some patients however do not present with the full spectrum of the disease. The hallmark of PJS is the presence of multiple hamartomatous polyps in the gastrointestinal tract. Most of these gastrointestinal polyps are found in the small intestine. However they can also be found in the stomach and also the large intestine.1 A diagnostic criteria was proposed by Giardello et al.,9 which required histopathological confirmation of hamartomatous gastrointestinal polyps and also two of the following features: small bowel polyposis, positive family history and pigmented skin or mucosal brown macules. PJS shows an autosomal dominant pattern of inheritance, with both familial (80%) and sporadic (25%) type of transmissions. It has been found that two independent groups of investigators have identified the gene mutation possible for PJS.14,15 The gene was localized to the chromosome 19p34-p36 and is known as STK11, which is a serine-threonine kinase involved in the growth control regulation. However, not all patients with PJS have a mutation in this gene. In a few families, mutations of chromosomes 6q and 19qhave also been seen.16 In the present case report, however the diagnosis was confirmed because of the hamartomatous small intestinal polyps and the mucocutaneous pigmentation over the perioral region and the buccal mucosa. There was no family history present of Peutz-Jeghers syndrome, in the present case, which was suggestive of a new sporadic mutation. The complications which can be caused by these polyps, include colicky abdominal pain, bleeding and bowel obstruction which can be due to intussusception. In our case report, colicky abdominal pai which was persistent was seen along with intussusception which was the main cause. The polyp formed the lead point of the intussusception in our case report. The commencement of abdominal symptoms in patients with PJS can vary from as early as the first year of life till that of 40 years of age.17 It has been observed that by the age of 10 years, about 30% of the patients of PJS, will require a laparotomy.18 Patients of PJS are prone to the risk of many extra-intestinal tumors as well, which include testicular Sertoli cell tumors, ovarian tumors like sex cord tumors with annular tubules, granulosa theca cell tumors, cystadenomas, breast tumors such as carcinoma breast, papilloma with squamous metaplasia, cholangioma, pancreatic adenocarcinoma, adenoma malignum, bronchial carcinoids and papillomas in the bladder and pelvis.19 If the polyps are symptomatic or are of a significant size (greater than 1.5cm in diameter) a laparotomy with enteroscopy is warranted. In our case report, the size of the polyp was about 3cm, for which a laparotomy was performed, and the polyp was excised through an enterotomy. Recently, instead of segmental resection of the bowel, intraoperative endoscopy and endoscopic polypectomy have been recommended. Endoscopic screenings, periodically every 2 years should be advocated.18 In the near future, the new mouth to anus (M2A) capsule endoscopy, will probably become the most useful screening tool for patients suspected for Peutz-Jeghers syndrome.
REFERENCES
Policy for Articles with Open Access
|
|

 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.